写在前面
运动障碍疾病曾被认为是神经病学的主要诊断分支,主要依靠现象学进行诊断,现已发展成为神经病学最具治疗导向的领域之一。系统的学习运动障碍疾病的治疗非常必要。
《Neurologic Clinics》杂志以“运动障碍疾病的治疗”为专题,邀请了最好的专家来提供全面、公平和权威的综述。这些综述用了很多的图片和表格阐述此类疾病的治疗方法,以提高每篇文章的临床和科学价值。非常感谢《Neurologic Clinics》的工作人员和撰写综述的专家教授们,本期我们团队翻译《Neurologic Clinics》杂志的运动障碍性疾病第七篇,
威尔逊病的概述和管控方法,仅供大家学习。
关键点
1.威尔逊病是一种可治疗的铜代谢遗传病,会导致铜沉积过多,并且会影响多种器官系统(最常见的是肝脏和大脑)的功能。
2.威尔逊病的初筛实验室检查应包括血清铜,血清铜蓝蛋白和24小时尿铜。 尽管基因检测可以确诊,但其他检测(例如脑部MRI和肝活检)可能会有所帮助。
3.威尔逊病的血清铜水平降低。4.目前的治疗方法包括限制饮食中的铜摄入量,服用锌剂抑制肠道对铜的吸收,螯合疗法从组织中去除铜以及对症治疗。早期发现,积极治疗预示着会有更好的预后。
威尔逊病是为数不多的运动障碍性疾病之一,目前存在可以改变疾病进展的疗法。该疾病主要是由肝脏和大脑中的铜超载所引起的,而铜超载是由ATP7B基因的遗传突变引起的铜排泄减少所致。铜过多会导致多种临床表现,包括神经系统症状,急性或慢性肝衰竭和/或精神症状。由于它是一种罕见的常染色体隐性遗传病,表现多种多样,因此通常会延误诊断和治疗,这是不幸的,因为它是可以治疗的。本文回顾了威尔逊病的临床表现,流行病学,遗传学,病理生理学,诊断和治疗。鉴于目前尚无公认的治疗方法,我们将根据我们的经验提供威尔逊疾病的管理方法。 临床表现威尔逊病于1912年由塞缪尔?亚历山大?金尼尔?威尔逊博士首次描述。在他发表在《Brain》上的文章中,他描述了几例与肝硬化相关的进行性神经功能障碍,并最终死亡。他提出这种疾病可能被称为“渐进性豆状核变性”,尽管最终以他的名字被称为威尔逊病。威尔逊病的许多核心特征已在他的第一篇文章中进行了描述,最突出症状包括神经功能障碍,肝硬化和精神症状,尽管目前知道还涉及其他系统。威尔逊病患者出现的症状有很大的变异性,因此有时被称为伟大的伪装者。必须记住,许多病例系列可能存在很大的转诊偏倚,因此神经系统,精神症状和肝病表现的频率差异很大。在印度对282例威尔逊病患者的一项研究发现,仅15%的患者出现肝病症状,69%的患者出现神经系统症状,4%的患者出现肝脏和神经系统症状,2%的患者仅出现精神症状。其他研究的估计值也有所不同,肝病医生报告的肝脏表现高达68%。精神症状通常在疾病早期没有得到重视。 发病年龄威尔逊病通常儿童期和成年期起病。发病的最常见的年龄为10到20岁,但也可以的5岁之前和70岁后发病。在一项研究中,经基因证实的1223名威尔逊病患者中有3.8%在40岁或更晚时出现症状。因此,尽管大多数患者都是早年患病,但仍可以在中年患者的鉴别诊断中考虑威尔逊病。有几项研究表明,以肝脏表现为主的患者往往比神经系统表现的患者年轻,尽管两者均可发生在生命的早期和晚期。 神经系统症状威尔逊病的神经系统症状多种多样,但大多数是指锥体外系功能障碍。威尔逊最初将神经系统症状描述为单纯的锥体外系症状,不自主运动,震颤,肌张力障碍,吞咽困难和构音障碍。这种描述仍然成立,但威尔逊病的神经系统症状包括构音障碍,肌张力障碍,步态异常,震颤,帕金森样表现,舞蹈病和癫痫发作(很少)。根据研究的不同,这些症状的发生频率有所差异。对几个独立病例系列的回顾表明,构音障碍是最常见的神经系统症状,其次是步态异常、小脑共济失调、肌张力障碍、帕金森样表现等,如表1所示。威尔逊病的肌张力障碍可以是局灶性,节段性或全面性,虽然面部表情的局限性肌张力障碍导致不自主的微笑被称为痉笑,相当常见于Wilson病。经典的威尔逊病中曾描述过一种扑翼样震颤,尽管在静止,姿势或动作时也可能出现震颤。威尔逊病中的肌张力障碍可以是局灶性,节段性或全身性,由于面神经局灶性肌张力障碍而引起的引起非自愿微笑的面部表情被称为痉笑,这在威尔逊病中相当普遍。尽管震颤可以在静止,姿势或动作时出现,但威尔逊病的经典震颤是扑翼样。
|
表格1基于4个大型独立病例研究的神经系统症状的总结
|
|
发病时的神经系统临床表现
|
病人(%)
|
|
|
构音障碍
|
46–97
|
|
|
步态异常/共济失调/小脑
|
28–75
|
|
|
肌张力障碍
|
38–69
|
|
|
帕金森症
|
12–58
|
|
|
姿势性震颤
|
55
|
|
|
吞咽困难
|
50
|
|
|
舞蹈症/徐动
|
6–30
|
|
|
癫痫发作
|
6–28
|
|
|
静止性震颤
|
4
|
|
肝功能不全威尔逊病的肝功能不全有一系列症状,从无症状的肝酶水平升高到暴发性肝衰竭。在疾病早期,通常转氨酶轻度增加,然后发展为慢性活动性肝炎,然后是纤维化,然后是肝硬化。肝硬化开始可能可以代偿,后期发展为伴有腹水,凝血功能障碍,静脉曲张和肝性脑病的失代偿性肝硬化。最常见的症状是黄疸,恶心和呕吐,发生率在37%至44%,其次是腹水,发生率在23%至36%,肝脾肿大的发生率在16%至29%。就诊时出现肝硬化预示着更高的死亡风险。有趣的是,威尔逊病患者肝细胞癌的风险似乎较低。 精神症状精神方面的临床表现通常是威尔逊病的首发症状,但是这些症状在神经或肝病症状发展之前通常不会诊断为威尔逊病。威尔逊在对这种疾病的最初描述中描述了12例中的8例存在“情绪化”。精神症状可能会有所不同,但情感障碍比精神病更为常见,且常见的精神症状包括人格改变,抑郁,认知变化和焦虑。人格改变可以表现为激进、脱抑制或强迫症。两个大型病例研究表明,许多患者在就诊时都有精神症状。一个回顾性病例中有64.8%的患者出现精神症状。在对195位患者的另一项研究中,有51%的患者有精神症状,而20%的患者在诊断出威尔逊病之前曾看过精神病医生。据估计,首发症状为精神症状的患者诊断延迟时间约为28个月,这远大于威尔逊病的其他临床症状,后者在另一项研究中平均约为12个月。这一发现不足为奇,因为在这个年龄段的人中抑郁很常见,轻度的神经系统副作用(例如震颤)可能是药物副作用导致。 眼科疾患Kayser-Fleischer环的首次描述在1902年和1903年,先于威尔逊病的最初描述。最初有关于它与威尔逊病的相关性有一些争论,但是一些研究已经确定,在90%至99%的神经系统症状病例中,存在K-F环。但是,只有超过一半的肝脏表现患者有K-F环。 K-F环的存在代表铜的沉积,最好由经验丰富的眼科医生进行裂隙灯检查来确定。但是,最近的一项研究还表明,某些裂隙灯检查正常的Wilson病患者显示光学相干断层扫描异常,这表明角膜的成像比传统的检测铜沉积更为敏感。 其他临床表现威尔逊病可能会影响其他几个器官系统,包括血液系统(贫血和血小板减少症),肾,内分泌和心脏系统。Coombs阴性的溶血性贫血和由溶血引起的黄疸可独立于肝病而发生,尽管这种症状很少出现。肾脏可能与威尔逊病有关,又独立于肝病,目前有肾小管酸中毒和氨基酸尿的报道。对骨和关节的影响可能表现为关节痛,有些患者被误诊为类风湿关节炎。有多种内分泌症状,包括发育不良和青春期疾病,甲状旁腺功能低下,痛经和不育。此外,还发现患有威尔逊病的患者在心电图和舒张功能障碍方面有轻度改变。 遗传学和流行病学威尔逊病是一种常染色体隐性遗传疾病,尽管直到1985年才发现其与13号染色体相关。1993年,两个独立的小组描述了威尔逊病患者ATP7B基因的突变。之后,在ATP7B基因出现了超过500个突变 ; 但是,还有很多东西要进一步研究。个体基因突变与威尔逊病的不同表现没有可靠的关联,尽管有证据表明,与错义突变相比,截短突变可能与更早的发病相关,而具有移码突变的患者可能更容易出现神经系统症状。有趣的是,有一些病例报告,单卵双胞胎的威尔逊病在表型上不尽相同。这一发现强烈表明,至少必须有一定程度的环境和表观遗传因素促成威尔逊疾病。但是,在鉴定可能修饰威尔逊病表型的其他遗传基因方面,成功涉及的基因有限,涉及到的几个基因包括甘油三酸酯代谢,脂质代谢和抗氧化途径。认为有所作为的环境因素包括性别、铁和饮食因素。 威尔逊病是一种罕见的疾病,尽管患病率因研究而异,被广泛引用的患病率为1/30000。中国和法国的最新研究表明,患病率分别为1/56000和1/66000。携带致病突变的个体的频率高于预期。这一发现已在法国,英国和中国的多个人群中得到证实。此外,一项研究表明,威尔逊病患者的后代中发生威尔逊病的频率高于预期。因此,关于威尔逊病的真正患病率以及在流行病学研究中是否存在未被确诊且未被发现的威尔逊病患者存在一些争议。另外,可能存在不完全的外显率,并且在文献中至少有1例患者的ATP7B基因携带2个突变并伴有肝炎,但没有任何铜代谢改变的证据。 生理学铜是许多酶的辅助因子,是正常代谢所必需的因子。例如,细胞色素C氧化酶是参与线粒体呼吸链的终端酶,铜作为辅助因子来完成这一过程。人们在饮食中每天消耗约1毫克的铜,并经胆汁排泄90%的未使用的铜,这些铜最终会由粪便排出,其余部分则由尿液排出。在威尔逊病中,患者无法有效地排泄胆汁中的铜,因此会在肝脏和大脑中积聚。 威尔逊病是由ATP7B基因的缺陷引起的,该基因编码铜转运P型ATP 酶。该蛋白的功能是将铜加载到高尔基体中的铜蓝蛋白酶上,从而变成肝细胞中的铜蓝蛋白。通常,铜蓝蛋白会与铜结合排泄到胆汁中,这是从体内清除多余铜的主要途径。在威尔逊病中,ATP7B蛋白的功能异常导致无法将铜加载到高尔基体中的铜蓝蛋白酶上,因此无法将铜适当地排泄到胆汁中(图1)。结果,铜在肝细胞中积累,随后渗入血液。铜存在于血液中的两个池中:铜蓝蛋白结合(85%–95%)和非铜蓝蛋白结合或游离铜(5%-15%),它们与白蛋白松散结合,可被细胞摄取或参与新陈代谢。前铜蓝蛋白是未结合铜的铜蓝蛋白,在血流中迅速降解,导致威尔逊病中血清铜蓝蛋白水平低。由于大多数血清铜通常与铜蓝蛋白结合,而威尔逊病中铜蓝蛋白水平低,因此该病中血清总铜水平低,游离未结合铜水平增加。铜毒性被认为是由氧化应激和自由基的产生介导的,自由基的产生是由参与代谢反应的游离铜介导的。最终,铜的毒性导致肝和神经功能障碍。对威尔逊病患者骨骼和血液问题的病理生理学知之甚少。
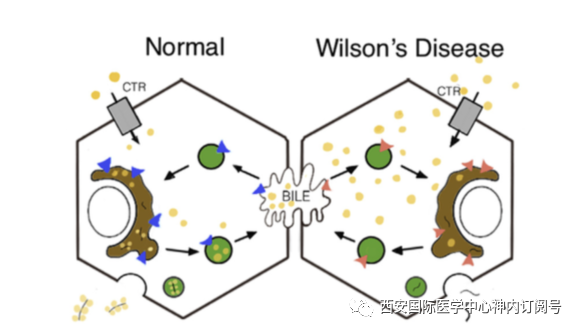
图1.在正常新陈代谢中(左图),铜(金点)通过铜转运蛋白进入肝细胞胞质,并由ATP7B基因编码的ATP酶(蓝色)进入高尔基体(棕色)。然后,铜通过囊泡运输,并以游离状态分泌到胆汁中或结合血浆蛋白(铜蓝蛋白)中。有缺陷的 ATP酶(红色)导致铜进入高尔基体和囊泡的运输受损,并减少了向胆汁的分泌。细胞内铜水平增加,导致肝细胞死亡和铜渗入血浆。在威尔逊病中,绝大部分前铜蓝蛋白未与铜结合,并被分泌到血浆中。然后它迅速降解,导致铜蓝蛋白水平降低。 诊断实验室检查一些实验室检查有助于Wilson病的诊断,在此进行总结,并附上诊断标准。 1.铜蓝蛋白大多数威尔逊病患者的血清铜蓝蛋白水平较低。众所周知,其他情况也可能导致铜蓝蛋白水平降低,包括ATP7B杂合突变个体,以及影响蛋白质丢失(例如肾病系统)或蛋白质合成(例如末期肝脏疾病,营养不良)的其他疾病。另外,急性时相反应或其他激素异常(例如怀孕和服用口服避孕药),铜蓝蛋白水平也可能增加。研究已经表明,铜蓝蛋白水平低(<20 mg / dL)诊断威尔逊病的敏感性为80%到99%。在中国对血清铜蓝蛋白的一项大型研究发现,铜蓝蛋白水平低于20 mg / dL对Wilson病的敏感性为99%,特异性为80.9%,但低于15 mg / dL的敏感性为95%特异性更高为95%。此外,对于有肝功能表现的患者,血浆铜蓝蛋白水平低可能无法诊断威尔逊病,一项研究发现,在肝病背景下铜蓝蛋白水平低对患者的阳性预测价值为5.9%。因此,诊断威尔逊病还需要其他因素,例如临床表现和其他实验室检查。2.血清铜和游离铜血清铜水平是血清中铜的总量,无论它与蛋白质(铜蓝蛋白或白蛋白)结合还是未结合(游离)。由于威尔逊病中铜蓝蛋白水平低,并且铜主要由血清中的铜蓝蛋白携带,因此尽管该病是由铜超负荷引起的,但威尔森病患者的血清总铜水平通常较低。血清铜本身对疾病不具有指示性及作用,但是游离铜是疾病的标志。尽管某些专门的实验室能够直接检测游离铜水平,但这种方法尚不广泛,因此通常通过血清总铜水平(ug / dL)减去铜蓝蛋白水平(mg / dL)乘以3来计算游离铜水平,以估算非铜蓝蛋白结合铜的水平。正常的游离铜水平低于10 ug / dL,在威尔逊病中通常高于该水平。但是,由于游离铜水平是一个间接值,因此它可以为负,并且并不总是可靠的。3.尿铜尿排泄铜是铜排泄的重要措施。如前所述,通常铜主要通过胆汁排出。在威尔逊病中,胆汁铜排泄减少,结果尿铜排泄增加。为了准确解释尿铜,患者必须带回家一个无铜的容器以收集尿液整整24小时。该要求在儿科患者中可能具有挑战性,并且如果未正确收集尿液,可能会导致假性降低。在威尔逊病中,成人24小时尿铜水平通常超过100 ug。如果尿铜水平轻度升高并且存在威尔逊病的问题,则可以在青霉素胺之前和之后测量尿铜,以评估青霉素胺后尿铜水平是否增加(每12小时500mg),如果组织中存储的铜过多,则可以诊断。4.其他的血液检查肝功能检查,包括天冬氨酸转氨酶(AST),丙氨酸转氨酶(ALT),γ-谷氨酰转移酶,胆红素,碱性磷酸酶和凝血酶原时间,可以帮助衡量肝功能。全血细胞计数可显示Coombs阴性溶血性贫血,血小板减少症等。5.头颅MRI对于那些有神经系统症状的患者,脑部MRI有助于确定与威尔逊病相符的发现,并排除神经系统症状的其他原因。尽管认为大熊猫脸征(由中脑中的T2信号增加)是威尔逊病的典型表现,但在威尔逊病中可能会发生多种MRI变化。一项回顾性研究对100例早期发作的锥体外系疾病患者与56例威尔逊患者进行了比较,发现大熊猫脸征出现在14%的威尔逊病患者和0%的其他儿科锥体外系,因此是相当特异的,但并不敏感。其他MRI表现包括基底节,丘脑,脑桥和白质信号改变及萎缩的。大多数接受治疗患者,MRI病变可逆,并且1项研究显示其与临床改善相关,尽管发生广泛变化的患者改善的可能性较小。6.肝活检肝活检是威尔逊病诊断的一部分,特别是对于有肝脏表现的患者。 总肝铜是肝活检中最有用的诊断方法,当肝铜水平高于250 ug / g时,其总灵敏度估计为83%,如苏丹红等染色可检测到溶酶体铜的异常存在,但由采样误差引起的假阴性仍然是使用肝活检诊断威尔逊病的重要问题。7.基因检测ATP7B基因检测被认为是诊断威尔逊病的金标准。但是,由于已描述的遗传突变的变异性,寻找常见的多态性会错过可能的致病突变,因此建议对整个基因进行直接测序。 诊断标准2003年,欧洲肝病研究协会(EASL)提出了诊断威尔逊病的诊断标准。这些标准综合考虑了临床和实验室检查结果,表2进行了总结。该标准结合了临床检查和实验室检查的结果,并在至少达到4分后才能确定诊断。这些标准用于临床研究。但是,它们对于医师试图确诊威尔逊病也很有帮助,尤其是在难以获得基因检测的情况下。
|
表格2威尔逊病的诊断标准
|
|
临床或实验室表现
|
|
评分
|
|
K-F环
|
存在不存在
|
20
|
|
神经系统症状或MRI发现
|
严重轻度缺乏
|
210
|
|
血清铜蓝蛋白水平(g / L)<0.1 2
|
<0.1 20.1–0.2 1正常(>0.2)
|
210
|
|
24小时尿铜>正常上限的2倍
|
>2倍正常值上限1-2倍正常值上限正常正常,但D-青霉胺治疗后> 5倍正常值上限
|
2102
|
|
Coombs阴性溶血性贫血
|
存在不存在
|
10
|
|
肝总铜水平(umol / g)> 5倍于正常上限
|
>5倍正常值上限(>4)增加(0.8–4)正常(<0.8)存在罗丹宁阳性颗粒
|
21-11
|
|
两个染色体上都存在遗传突变
|
存在于两条染色体上存在于1条染色体上不存在
|
410
|
|
总分
|
诊断成立可能诊断,需要更多检查诊断可能性不大
|
432或更少
|
管理治疗的总体目标是通过平衡铜的摄入和排泄来建立正常的铜稳态。对于铜过载的患者,治疗的目标是净铜负平衡,可以通过螯合疗法增加排泄,以及通过锌减少机体对铜的吸收和减少饮食摄入来实现。一旦建立了适当的平衡,通常仅使用锌和低铜饮食即维持平衡,但有时需要进行持续的螯合疗法。 药物治疗多种药物被批准用于治疗Wilson病,1种药物目前正在进行III期临床试验。药物的机理,副作用和效力各不相同,表3对此进行了总结。D-青霉胺和曲恩汀都通过与铜结合而充当螯合剂,然后在尿液中排出,从而促进铜排泄。相反,锌盐通过诱导肠内和肝脏中内源性铜螯合剂金属硫蛋白的表达而抑制了饮食中铜的吸收。双胆碱-四硫代钼酸盐目前正在研究中,具有新颖的作用机理,其作用是将游离铜络合到白蛋白上,螯合游离铜参与代谢反应。以前曾研究过类似的药物四硫钼酸铵,具有抗铜作用,但对实际应用而言太不稳定了。这些药物在降低体内总铜水平方面的效果也有所不同,其中D-青霉胺对铜的外排作用最大,而锌盐的作用最弱。对接受曲恩汀和D-青霉胺治疗的患者进行的一项大型回顾性研究支持这两种药物具有相似的疗效,但D-青霉胺组的副作用增加。但是,比较抗铜药物疗效的研究质量仅限于较小的非随机试验。meta分析确实支持D-青霉胺优于安慰剂,但比较锌与D-青霉胺的研究存在混杂因素。当需要进行螯合治疗时,作者通常更喜欢从曲恩汀开始而不是D-青霉胺治疗,因为它的副作用更少。 在选择威尔逊病的治疗方法时,潜在的神经系统恶化是另一个重要考虑因素。许多研究中已经描述了开始螯合治疗后早期神经系统症状恶化的频率发生在不到10%至50%的患者中。不同神经螯合剂之间是否存在恶化的频率差异仍不清楚。曲恩汀治疗的患者中血清游离铜水平升高先于神经系统恶化,并且已经提出螯合增加了游离铜,其可产生自由基并有助于进一步的破坏。因此,如果患者有神经系统症状且需要密切监测,则应以低剂量开始螯合治疗。如果确实发生神经功能恶化,则应减少甚至停止使用螯合剂,并应使用锌疗法。这种情况通常可能是一个难题,因为目标是尽可能快地降低大脑和肝脏中的铜水平以减少永久性损害,但是与传统螯合剂螯合得太快会造成额外损害。因此,从理论上讲,四硫代钼酸盐络合游离铜的新机理在理论上是有吸引力的,并且最近的II期试验支持了神经系统恶化的低比率,尽管还需要进行更大规模的研究。
|
表格3威尔逊病的药物
|
|
药物
|
作用机制
|
剂量
|
副作用
|
|
D-青霉胺
|
螯合铜并促进尿排泄
|
最高1 g / d,分为2-4次必须在饭前30分钟或饭后2小时服用
|
发烧,皮疹,贫血,骨髓抑制,淋巴结肿大,狼疮样综合征,神经系统症状恶化
|
|
曲恩汀
|
螯合铜并促进尿排泄
|
最高1 g / d,分为2-4次必须在饭前30分钟或饭后2小时服用
|
蛋白尿,骨髓抑制,自身免疫反应,神经系统症状恶化
|
|
锌盐
|
在胃肠道上皮中诱导金属硫氨酸并抑制铜的吸收
|
50 mg TID必须在饭前或饭后至少1小时服用
|
胃肠道不适
|
|
双胆碱-四硫代钼酸盐(III期临床试验中)
|
将游离铜与白蛋白结合
|
15 mg/d
|
转氨酶升高,骨髓抑制
|
肝移植肝移植是肝病患者的有效疗法。将正常运行的肝脏移植到Wilson病患者中,基本上可以恢复正常的铜排泄以及正常的肝功能。但是,肝移植是一个复杂的过程,需要终生免疫抑制,通常仅用于急性肝功能衰竭或失代偿性肝硬化患者。 饮食遵循低铜饮食是一种常识性的生活方式改变,应该可以减少人体的总铜负荷。一般而言,建议患者的总铜消耗量应少于1 mg / d,这可以通过避免食用铜含量最高的食物(例如贝类和肝脏)并食用其他富含适量铜的食物来实现(即巧克力,坚果,干果,豆类和蘑菇)。如果水来自铜管道,则水中的铜含量可能很高。尽管自来水冲洗管道会大大降低铜水位,但大多数水过滤器不会去除铜。纯净水或蒸馏水几乎不含铜。避免使用铜锅。没有直接证据表明低铜饮食可以治疗威尔逊病,但是在合理范围内减少摄入是合乎逻辑的。
症状管理尽管很多注意力都集中在抗铜疗法上,但是症状管理也是药物治疗的重要组成部分。对症处理肝脏并发症如食管静脉曲张和腹水,类似于其他原因的晚期肝病。对于神经系统症状,可以尝试抗帕金森症的药物,例如卡比多巴/左旋多巴,但不一定有效。可以尝试治疗肌张力障碍的方法,包括抗胆碱能药物,如苯海索或肉毒杆菌毒素局部注射,治疗局灶性肌张力障碍。已经尝试过脑深部电刺激丘脑腹中间核治疗震颤和苍白球内侧部治疗肌张力障碍,在少数患者中取得了不同的成功。此外,采用多学科方法,进行言语治疗缓解言语障碍和吞咽困难,以及物理治疗和职业治疗也可能会有帮助。可以采用五羟色胺再摄取抑制剂或神经安定药物治疗精神症状,有证据表明,威尔逊病患者对神经安定药更为敏感。 预防一旦患者被诊断为威尔逊病,就应该对兄弟姐妹进行风险筛查。由于威尔逊病是一种常染色体隐性遗传疾病,因此患者的兄弟姐妹患该病的风险为25%。兄弟姐妹,无论是否有症状,应进行肝功能检查,神经系统检查,眼科检查和铜代谢功能检查(铜蓝蛋白,24小时尿铜)或基因检查进行筛查。如果已知特定的遗传突变,则可以测试同胞的多态性,或者可以对ATP7B基因中的多态性进行单倍型分析。另外,已经努力开发针对新生儿的人口筛查测试,以检测症状前的威尔逊病。尽管对铜蓝蛋白的初步研究未能成功,但一项量化血清中ATP7B蛋白含量的新测试有望成为一种合适的筛查工具。 展望在医学治疗技术发展之前,威尔逊病是不可避免的致命性疾病。早期诊断很重要,并且已证明与降低死亡率和需要肝移植有关,并且预期寿命接近普通人群。此外,接受治疗的威尔逊病患者的健康相关生活质量与普通人群相似。但是,依从性是影响预后的重要因素,有报道称,以前接受过药物控制的患者不依从导致死亡。鉴于症状的可逆性,大多数证据表明,神经系统和肝脏症状经过适当的治疗大多数患者能得到改善,肌张力障碍可能是最有可能对治疗有反应的神经系统症状,精神症状也可通过抗铜治疗改善,尽管治疗2年后可能会出现平台期。尽管大多数患者对治疗的反应良好,但也有报道显示即使早期诊断和治疗,患者仍死亡,有一个概念即某些患者对传统的驱铜治疗没有效果。管理方法对于威尔逊病患者的治疗方法尚无共识,但总体方法有一些一致的观点。作者想分享我们威尔逊疾病中心的治疗方法。当对新诊断的患者开始抗铜治疗时,治疗的目标是达到铜负净平衡。但是,应根据临床情况评估降低铜水平的紧迫性。当症状已经出现时,就更迫切需要降低铜水平以最大程度地缓解症状,并防止疾病进一步发展,因此需要使用更有效的螯合药物。在无症状的患者中,降低铜水平的紧迫性较小,有可能单独使用锌和低铜饮食。 对于无症状的患者,通常每天空腹服用3次锌,每次50毫克,并同时低铜饮食。患者每3个月监测神经系统症状和肝功能检查以及尿铜排泄量。如果患者在任何时候出现症状,我们会添加螯合剂。如果尿铜水平比基线水平高,也可考虑在那时添加一种螯合剂。
对于有症状的患者,考虑到副作用,如果患者的医疗保险可用,我们通常会使用曲恩汀,但如果没有曲恩汀,则可以使用D-青霉胺。对于这两种药物,我们通常在空腹时以250 mg / d的剂量开始服用,然后每7到14天以250 mg的剂量递增,每天最多增加1000到1500 mg,分为2到3次。对于体重不超过45千克的儿童和成人,我们建议每天250毫克开始服用,最大总日剂量为20毫克/公斤体重,分2或3次。我们会密切监测神经系统症状,如果有证据表明症状加重,应减少剂量。螯合期间大约每3个月监测一次肝功能,全血细胞计数,血清铜和铜蓝蛋白以及24小时尿铜。我们发现24小时尿铜比血清中游离铜的水平(间接测量)更准确。一旦螯合开始,尿液中的铜含量会增加,但随着铜存储的耗尽,尿液中的铜含量最终会下降。一旦24小时尿铜水平降至100-200 ug / d(通常可能需要6到12个月),患者就会进入治疗的维持阶段。
在治疗的维持阶段,目标是维持净铜平衡。螯合治疗后,尽管有些患者仍接受低剂量螯合治疗,但许多患者仅靠锌和低铜饮食就能达到这一目的。如前所述,我们使用尿铜作为铜状态的标志,在维持阶段每6个月对尿铜进行监测,同时还要进行血清铜,铜蓝蛋白,全血计数和肝功能检查。尿铜含量增加100 ug/ d以上表明全身铜存储量增加,这反映了驱铜治疗依从差或不足,应通过强调依从性,增加后备螯合剂或增加螯合剂的剂量来解决这种情况,直到尿铜水平再次降至低于100 ug / d。由于威尔逊病的慢性特征以及依从性差导致的加重风险,我们每6个月持续监测实验室指标。
Caitlin Mulligan,Jeff M. Bronstein,Wilson Disease An Overview and Approach to Management,https://doi.org/10.1016/j.ncl.2020.01.005

 作者:西安国际医学中心
作者:西安国际医学中心
 时间:2020-07-23
时间:2020-07-23
 浏览:3585次
浏览:3585次




